








Optimizing protein structures with NOVA
One of the conclusions drawn early at the CASP meetings was that applying various force fields during refinement of template-based models tends to move predictions in the wrong direction - away from the experimentally determined coordinates. NOVA is an all-atom force field aimed at protein + nucleotide optimization in vacuo which has been specifically designed to avoid this problem. NOVA resembles common molecular dynamics force fields, but has been automatically parameterized with two major goals:
1) Not to make high resolution X-ray structures worse and
2) to improve homology models built by WHAT IF.
Force field parameters were not required to be physically correct, instead they were optimized with random Monte Carlo moves in force field parameter space, each one evaluated by simulated annealing runs of a 50 protein optimization set. Errors inherent to the approximate force field equation could thus be canceled by errors in force field parameters. When compared to the optimization set, the force field did equally well on an independent validation set and is shown to move in silico models closer to reality. It can be applied to modeling applications as well as X-ray and NMR structure refinement.
- A detailed description of NOVA can be found at:
Increasing the precision of comparative models with YASARA NOVA - a self-parameterizing force field.
Krieger E, Koraimann G & Vriend G (2002) Proteins 47, 393-402. - To download a PDF reprint, click here.
The NOVA force field has been relaunched in January 2012, with much higher performance (SIMD&multi core support), much higher accuracy (knowledge-based potentials), and automatic force field parameter assignment for organic molecules. More details about the improved NOVA force field can be found here.
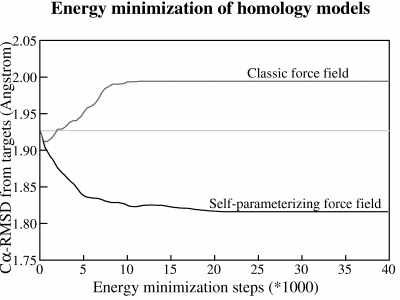
Figure 1: The average RMSD between models and targets during an extensive energy minimization of 14 homology models with two different force fields. Both force fields improve the models during the first ~500 energy minimization steps but then the small errors sum up in the classic force field and guide the minimization in the wrong direction, away from the target, while the self-parameterizing NOVA force field goes in the right direction. To reach experimental accuracy, the minimization would have to proceed all the way down to ~0.5Å which is the uncertainty in experimentally determined coordinates.